BCM is a rare genetic disease of the retina caused by genetic mutations on genes OPN1LW, OPN1MW and LCR. These genes are located on the X chromosome and encode proteins called photopigment, needed in the red and green cones to capture light.
On this page we will see what are the genes responsible for color vision, what are the genetic mutations that lead to disease and the history of these scientific discoveries of molecular genetics.
The BCM Families Foundation supports the research of Molecular Genetics on BCM and the creation of a BCM Patients Registry, in order to deepen the knowledge about the genes and the genetic mutations that cause disease.
Molecular genetics of human color vision
BCM is inherited from the X chromosome. As any other chromosome, X contains a long molecule of DNA, the chemical of which genes are made.
The X chromosome is composed of two arms, the upper is called p, the lower q.
The genes involved in the BCM are in position Xq28, at the end of the q arm.
In the figure below we see the X chromosome:
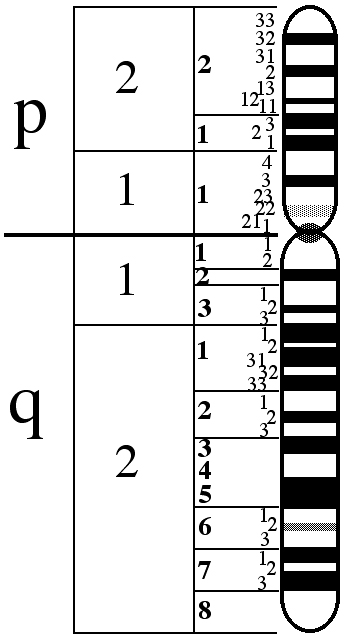
In the position Xq28 there are in order, the genes named LCR, OPN1LW and OPN1MW.
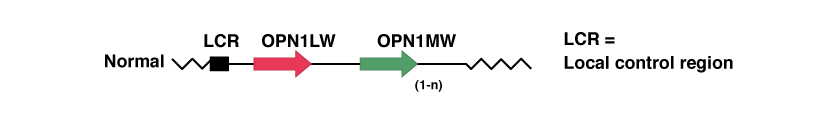
LCR is the Locus Control Region, and acts as a promoter of the expression of the two opsin genes thereafter. In the absence of this gene, none of the following two genes are expressed in the human retina. In addition, it ensures that only one of the two opsin genes (red or green) is expressed exclusively in each cone.
OPN1LW and OPN1MW are respectively the genes that contain the genetic code for protein opsin. These proteins constitute the photopigments for the capture of light, red (Long Wave) and green (Medium Wave).
Many people have several replicas of the gene for the green photopigment, OPN1MW. Only the first two genes, immediately after the LCR, (the red and the first green ones), are expressed in the retina. Approximately 25% of male Caucasians have a single OPN1MW gene, while 50% have two genes and the remainder have 3 or more genes.
In the following figure we see a representation of the opsin gene array in a normal case:
To learn more about these genes, please refer to the web site of the ‘National Center for Biotechnology Information’, NCBI, particularly to:
The gene responsible for the formation of the blue photopigment is in a position far away, on chromosome 7 and the gene responsible for the formation of rhodopsin (the rod photopigment) is located on chromosome 3:
In the following figure we see the opsin proteins, the blue ‘S’ (short), the green ‘M’ (medium) and the red ‘L’ (Long) one.
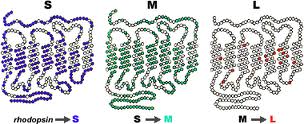
(OPSIN Genes – Picture is taken from handprint.com)
They take the form of a chain passing 7 times through a disk of the outer segment of a cone. The three proteins are very similar between them and, in particular, the M and L differ only in some elements that compose them. The two photopigments, red and green are in fact equal to 96%, while they have only a 46% similarity with the blue photopigment.
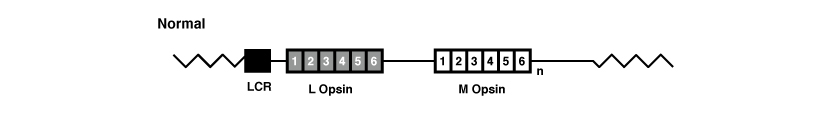
The genes OPN1LW and OPN1MW, like all genes, are formed by exons and introns. In particular both of these genes have six exons, referred to as 1 to 6.
(Picture is taken from Jessica C. Gardner, Michel Michaelides, Graham E. Holder, Naheed Kanuga, Tom R. Webb, John D. Mollon, Anthony T. Moore, Alison J. Hardcastle ‘Blue cone monochromacy: Causative mutations and associated phenotypesMolecular Vision 2009; 15:876-884).
Like all proteins, the opsin proteins are three-dimensional structures that need to perform a ‘folding‘ to assume their final three-dimensional shape. Some specific amino acids within the protein are responsible for the folding of the same.
The Genetic Mutations
There are many genetic mutations that can affect this group of genes, LCR, OPN1LW and OPN1MW.
Some mutations lead to conditions commonly called ‘color blindness’, having as its only effect the inability to distinguish certain colors.
Mutations that lead to the BCM to date identified are the following:
Large deletions
1. Deletion of the LCR, or deletion of the LCR and some or all of the exons of the gene OPN1LW.
This mutation is an absence of a large part of the genetic material. Since there isn’t the genetic code for LCR the two opsin proteins will not express and the cones will not have the red or green photopigments.
2. Intragenic deletion. This is a deletion of exons within the genes OPN1LW and OPN1MW or deletion of genetic material of the first and of the second gene.
Even this mutation is an absence of a large part of the genetic material.
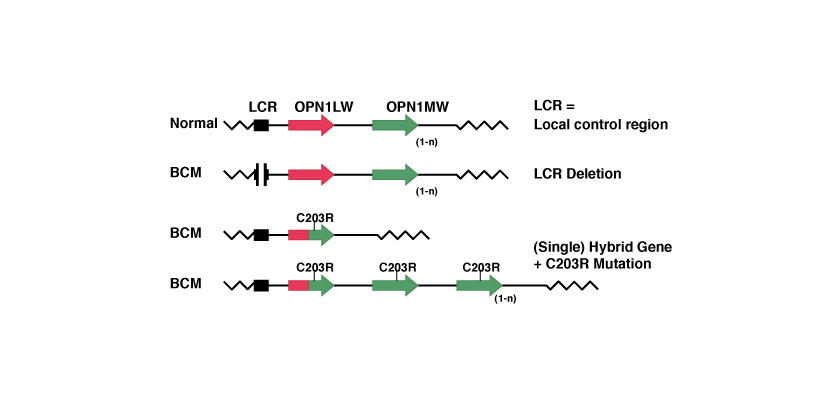
Mechanism in 2 steps with homologous recombination and inactivation.
In this case, due to the similarity between the two genes OPN1LW and OPN1MW, during a process of ‘homologous recombination’ one of the two genes is lost with the creation of an hybrid gene. Subsequently, a point mutation inactivates the remained gene.
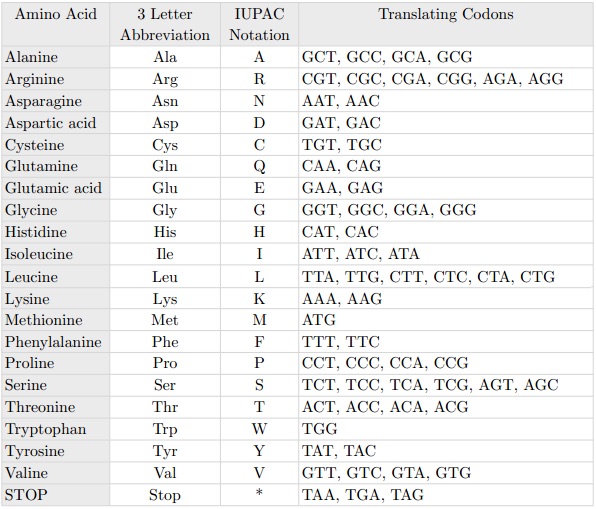
The point mutation best known is the so-called C203R. The name of the point mutations indicates the position at which mutation has occurred, in this case the amino acid position 203 and which has been replaced, in this case a C = Cysteine with an R = Arginine. At the level of codons this substitution is timely because it corresponds to replace thymine with cytosine in position 648, as we see from the following table:
The C203R mutation causes the opsin protein once formed to not fold into the proper three-dimensional form.
(Picture is taken from Jessica C. Gardner, Michel Michaelides, Graham E. Holder, Naheed Kanuga, Tom R. Webb, John D. Mollon, Anthony T. Moore, Alison J. Hardcastle ‘Blue cone monochromacy: Causative mutations and associated phenotypesMolecular Vision 2009; 15:876-884).
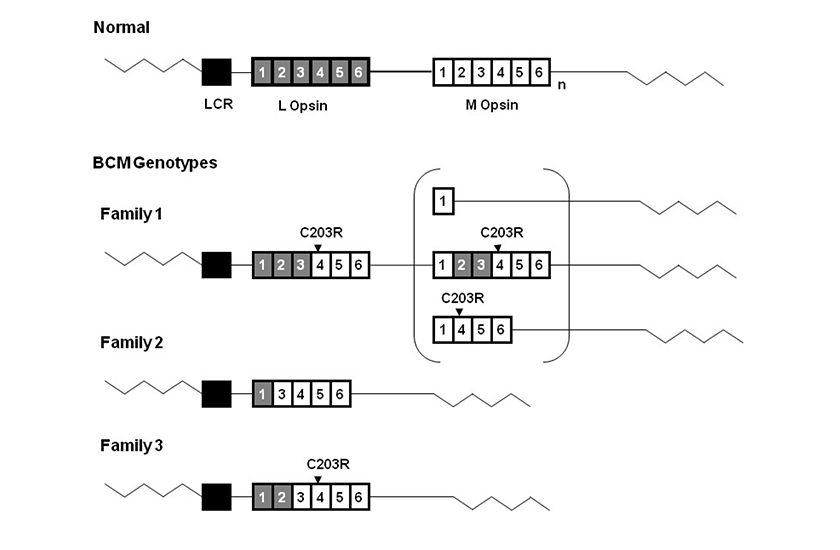
Diagram representing BCM genotypes of 3 British families. The wild type L-M opsin gene array is shown at the top of the figure. Gray boxes represent L opsin exons and white boxes represent M opsin exons.
Subscript n represents one or more M opsin genes. The black box represents the Locus Control Region, LCR. The LCR was present without mutation in all three families. The C203R point mutations detected in Family 1 and Family 3 are shown above the corresponding exons. Family 1 has an inactive hybrid gene followed by a second gene in the array. Three possible structures of this second inactive gene are shown in the bracket.
Family 2 has a single nonfunctional hybrid gene lacking exon 2. Family 3 has a single inactive hybrid gene.
Other point mutations are the P307L, and R247X. The last one replaces arginine with a stop codon at position 247 which prevents formation of the protein (nonsense mutation).
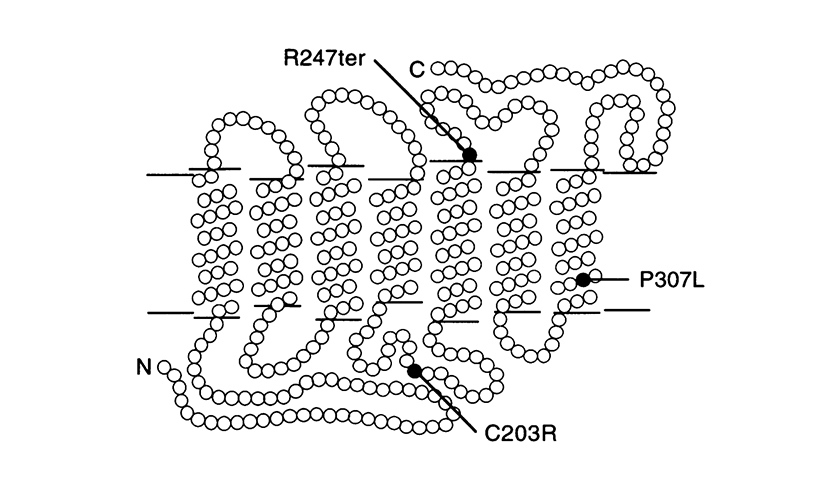
(The picture is taken from J. Nathans et al. Am. J. Hum. Genet. 53: 987-1000, 1993).
Model of the red and 5’ red – 2’ green hybrid pigments in the photoreceptor membrane showing the locations of point mutations identified in blue-cone monochromats. Each circle represent an amino acid. ‘N’ = amino-terminus and ‘C’ = car-body-terminus. The amino-terminus faces the extracellular space.
Other mutations
Other mutations on genes OPN1LW and OPN1MW that lead to the BCM are constituted by a set of point mutations called for example LIAVA. The BCM will be caused by the production of new hybrid gene, like in the previous case, from the homologous recombination of OPN1LW and OPN1MW. In this case exon 3 contains the following amino acids in the positions indicated: 153 Leucine, 171 Isoleucine, 174 Alanine, 178 Valine and 180 Alanine. This genotype has the abbreviated name LIAVA.
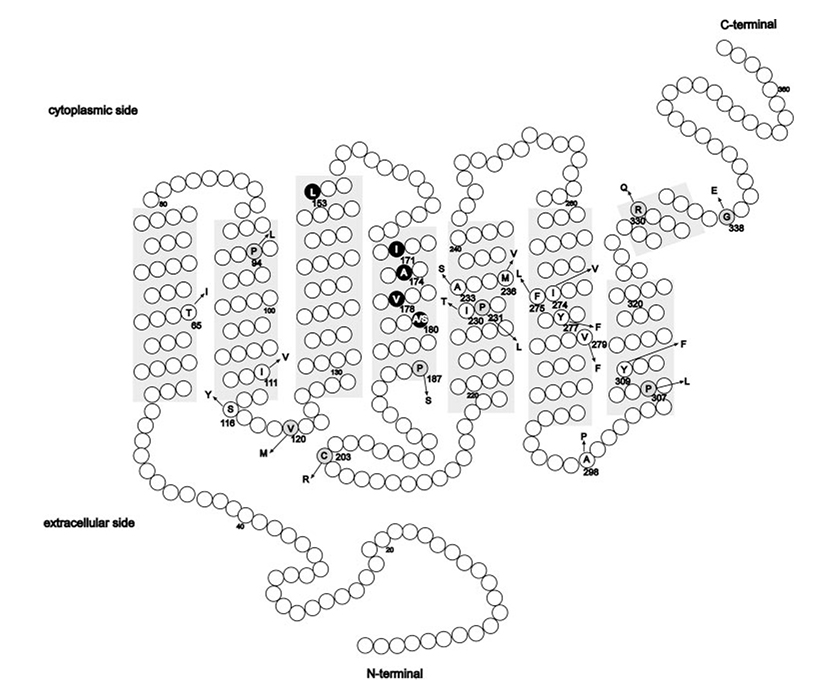
(Picture is taken from Mizrahi-Meissonnier L., Merin S., Banin E., Sharon D., 2010).
Location of amino acid alterations reported thus far in the L and M cone opsin genes. Shaded areas: the transmembrane domains. Circles: amino acid differences and known polymorphism with the more common amino acid (in a one-letter code); arrow: the amino acid change. The codon number is depicted for each change. Missense changes associated with a cone-opsin-related disease that are likely to cause protein dysfunction are on a gray background. The LIAVA haplotype is highlighted in black.
Nathans et al. 1993 | ||
Ayyagari et al. 2000 | ||
A.Reitner, L.T. Sharpe, E. Zrenner 1991 | ||
Mizrahi-Meissonnier L. et al. 2010 Neitz M. et al. 2004 Crognale MA. et al. 2004 |
Other diseases with genetic mutations on genes and OPN1LW OPN1MW
Another disease of the retina that is associated with the position Xq28 is the Bornholm Eye Disease (BED), with symptoms similar to those of the BCM. It is a very rare disease and it is stationary. For further information you can consult OMIM and the web site of University of Arizona.
Finally note there is also a particular mutation of the two genes OPN1LW and OPN1MW which causes a different disease from the BCM. This type of mutation is W177R and is a misfolding mutation that, if present on both opsin genes causes cone dystrophy with evidence of degeneration and cell death of the cones.
The History of the discovery of the genes of the BCM
Many researchers have contributed to discoveries about the genes involved in the BCM.
We recall the fundamental discoveries of Jeremy Nathans on the genes responsible for color vision:
Nathans, J., Thomas, D., Hogness, D. S. Molecular genetics of human color vision: the genes encoding blue, green, and red pigments. Science 232: 193-202, 1986. PMID:2937147.
Nathans, J., Piantanida, T. P., Eddy, R. L., Shows, T. B., Hogness, D. S. Molecular genetics of inherited variation in human color vision. Science 232: 203-210, 1986. PMID: 3485310 .
Nathans, J. Molecular biology of visual pigments. Annu. Rev. Neurosci. 10: 163-194, 1987. PMID: 3551758 .
Nathans, J. The evolution and physiology of human color vision: insights from molecular genetic studies of visual pigments. Neuron. 24: 299-312, 1999. PMID: 10571225 .
Deeb, S. S. The molecular basis of variation in human color vision. Clin. Genet. 67: 369-377, 2005. PMID: 15811001 .
In particular, the work that led us to understand the main causes of BCM and in particular the 2-step process with point mutation C2013R:
Nathans, J., Davenport, C. M., Maumenee, I. H., Lewis, R. A., Hejtmancik, J. F., Litt, M., Lovrien, E., Weleber, R., Bachynski, B., Zwas, F., Klingaman, R., Fishman, G. Molecular genetics of human blue cone monochromacy. Science 245: 831-838, 1989. PMID: 2788922.
Nathans, J., Maumenee, I. H., Zrenner, E., Sadowski, B., Sharpe, L. T., Lewis, R. A., Hansen, E., Rosenberg, T., Schwartz, M., Heckenlively, J. R., Traboulsi, E., Klingaman, R., Bech-Hansen, N. T., LaRoche, G. R., Pagon, R. A., Murphey, W. H., Weleber, R. G. Genetic heterogeneity among blue-cone monochromats. Am. J. Hum. Genet. 53: 987-1000, 1993. PMID: 8213841.
Reyniers, E., Van Thienen, M.-N., Meire, F., De Boulle, K., Devries, K., Kestelijn, P., Willems, P. J. Gene conversion between red and defective green opsin gene in blue cone monochromacy. Genomics 29: 323-328, 1995. PMID: 8666378.
An important work for the type of mutations ‘Deletion’ of the LCR or LCR and the gene OPN1LW is:
Ayyagari, R., Kakuk, L. E., Bingham, E. L., Szczesny, J. J., Kemp, J., Toda, Y., Felius, J., Sieving, P. A. ‘Spectrum of color gene deletions and phenotype in patients with blue cone monochromacy’. Hum. Genet. 107: 75-82, 2000. Hum Genet. 2000 Jul;107(1):75-82. PMID: 10982039.
For the Deletion intragenic the following works identified a case of BCM with the presence of only the gene OPN1LW (red) without the exon 4:
Ladekjaer-Mikkelsen, A.-S., Rosenberg, T., Jorgensen, A. L. ‘A new mechanism in blue cone monochromatism’. Hum. Genet. 98: 403-408, 1996. PMID: 8792812.
Reitner, A., Sharpe, L. T., Zrenner, E. Is colour vision possible with only rods and blue-sensitive cones? Nature 352: 798-800, 1991. PMID: 1881435 .
The Locus Control Region, and its role in the expression of opsin genes, was the result of the following works:
Lewis, R. A., Holcomb, J. D., Bromley, W. C., Wilson, M. C., Roderick, T. H., Hejtmancik, J. F. Mapping X-linked ophthalmic diseases: III. Provisional assignment of the locus for blue cone monochromacy to Xq28. Arch. Ophthal. 105: 1055-1059, 1987. PMID: 2888453 .
Lewis, R. A., Nathans, J., Holcomb, J. D., Bromley, W. C., Roderick, T. H., Wilson, M. C., Hejtmancik, J. F. ‘Blue cone monochromacy: assignment of the locus to Xq28 and evidence for its molecular rearrangement’. Am. J. Hum. Genet. 41: A102 only, 1987.
Wang, Y., Macke, J. P., Merbs, S. L., Zack, D. J., Klaunberg, B., Bennett, J., Gearhart, J., Nathans, J. ‘A locus control region adjacent to the human red and green visual pigment genes’. Neuron 9: 429-440, 1992. PMID: 1524826.
In particular, the role of LCR that allows the exclusive expression of a unique opsin (red or green) in each cone, was reported in the last publication.
For the study of the C203R mutation please see the following research publications:
Kazmi MA, Sakmar TP, Ostrer H. ‘Mutation of a conserved cysteine in the X-linked cone opsins causes color vision deficiencies by disrupting protein folding and stablilty’. Investigative Ophthalmology and Visual Science. 1997;38(6):1074–1081.
who understood the negative effects of this mutation on the folding of the opsin protein and:
Winderickx J, Sanocki E, Lindsey DT, Teller DY, Motulsky AG, Deeb SS. Defective colour vision associated with a missense mutation in the human green visual pigment gene. Nature Genetics. 1992;1:251–256. PMID: 1302020 .
who studied this mutation and its frequency of about 2% in people of Caucasian origin.
On rare mutations of the type LIAVA you can consult:
Carroll J1, Neitz M, Hofer H, Neitz J, Williams DR., ‘Functional photoreceptor loss revealed with adaptive optics: an alternate cause of color blindness.’ Proc Natl Acad Sci U S A. 2004 Jun. PMID: 15148406.
Mizrahi-Meissonnier L., Merin S., Banin E., Sharon D., ‘Variable retinal phenotypes caused by mutations in the X-linked photopigment gene array’. Invest. Ophthalmol. Vis. Sci. 2010 Aug;51(8):3884-92. PMID: 20220053.
Neitz M, Carroll J, Renner A, et al. ‘Variety of genotypes in males diagnosed as dichromatic on a conventional clinical anomaloscope’. Vis Neurosci. 2004;21:205–216. PMID: 15518190.
Crognale MA, Fry M, Highsmith J, et al. ‘Characterization of a novel form of X-linked incomplete achromatopsia’. Vis Neurosci. 2004; 21:197–203. PMID: 15518189.
Some ancinent initial research about BCM were:
Huddart, J. ‘An account of persons who could not distinguish colours’. Phil. Trans. Roy. Soc. 67: 260 only, 1777.
Sloan, L. L. ‘Congenital achromatopsia: a report of 19 cases’. J. Ophthal. Soc. Am. 44: 117-128, 1954. PMID: 13131176.
Alpern, M., Falls, H. F., Lee, G. B. ‘The enigma of typical total monochromacy’. Am. J. Ophthal. 50: 996-1012, 1960. PMID: 13682677.
Blackwell, H. R., Blackwell, O. M. Rod and cone receptor mechanisms in typical and atypical congenital achromatopsia. Vision Res. 1: 62-107, 1961. https://doi.org/10.1016/0042-6989(61)90022-0.
Fleischman, J. A., O’Donnell, F. E. Jr. ‘Congenital X-linked incomplete achromatopsia. Evidence for slow progression, carrier fundus findings, and possible genetic linkage with glucose-6-phosphate dehydrogenase locus’. Arch Ophthalmol 1981;99:468-472. PMID: 6971088.
Lewis, R. A., Holcomb, J. D., Bromley, W. C., Wilson, M. C., Roderick, T. H., Hejtmancik, J. F. ‘Mapping X-linked ophthalmic diseases: III. Provisional assignment of the locus for blue cone monochromacy to Xq28′. Arch. Ophthal. 105: 1055-1059, 1987. PMID: 2888453.
For the study of cone dystrophy, a degenerative disease caused by a point mutation on both genes OPN1LW and OPN1MW:
Gardner JC, Webb TR, Kanuga N, Robson AG, Holder GE, Stockman A, Ripamonti C, Ebenezer ND, Ogun O, Devery S, Wright GA, Maher ER, Cheetham ME, Moore AT, Michaelides M and Hardcastle AJ,’X-Linked Cone Dystrophy Caused by Mutation of the Red and Green Cone Opsins’.The American Journal of Human Genetics 87, 26–39, July 9, 2010. PMID: 20579627.
Here there are some review publications that illustrate the topic:
Neitz J., Neitz M. ‘The genetics of normal and defective color vision’. 2011 Review. Vision Research. PMID: 21167193.
Deeb, S.S. ‘Molecular Genetics of colour vision deficiencies’. Clinical and Experimental Optometry 87.4 – 5 July 2004. PMID:15312026.
